JMod
Joint modeling of mass spectra for empowering multiplexed DIA proteomics.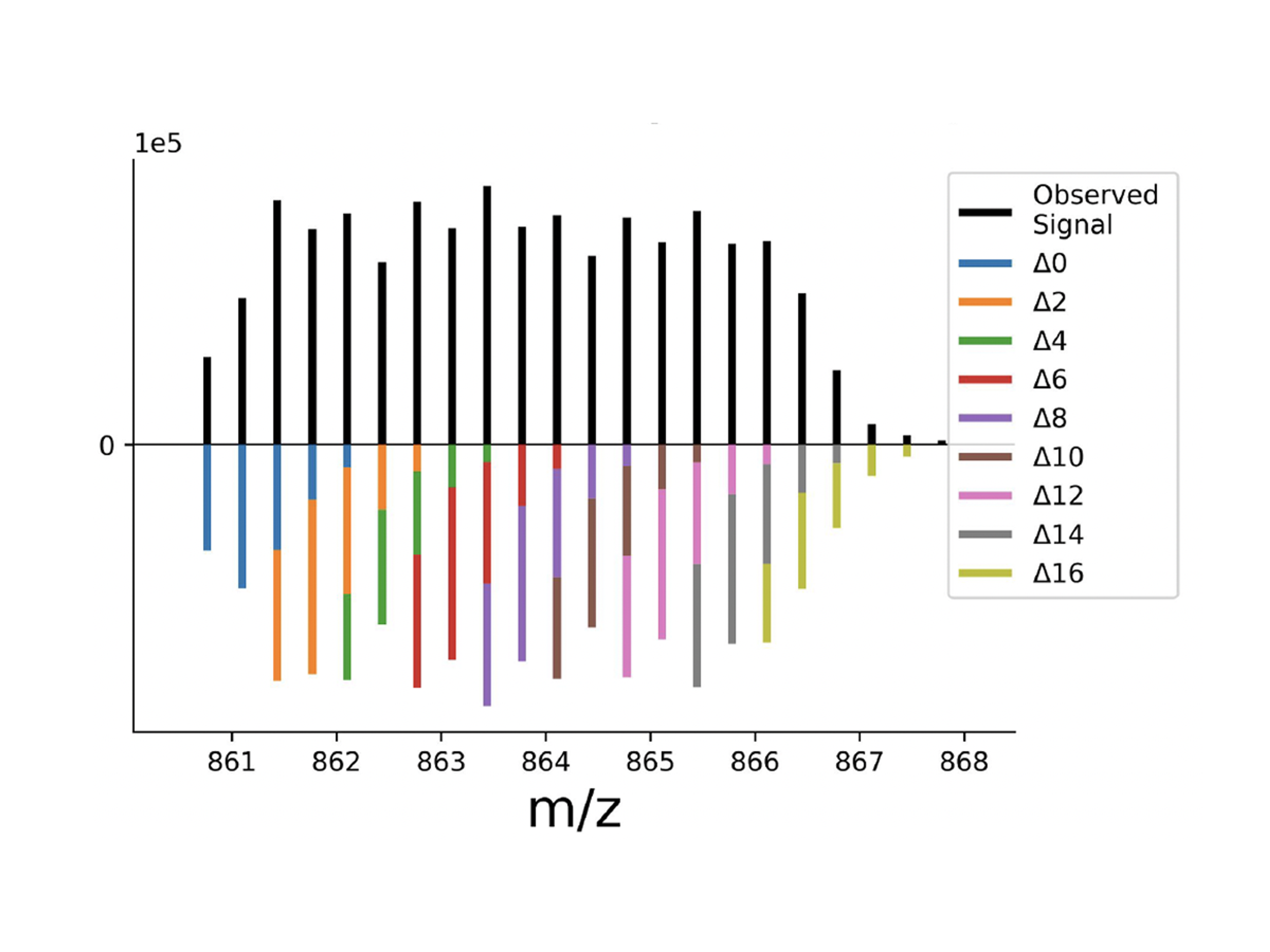
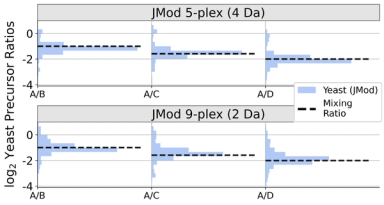
To meet increasing data complexity inherent to multiplexing, here we introduce JMod, an open-source platform that models peak superpositions to enable increased DIA multiplexing. The model is designed for deconvolving the structured overlap that arises in both MS1 and MS2 spectra from DIA acquisition of samples multiplexed by mass tags (plexDIA), by sample-specific retention time offsets (timePlex), or by both combined. JMod uses a linear superposition model to leverage structure in the data created by multiplexing to improve sequence identification and to resolve quantitative differences between overlapping precursor and fragment ions. Moreover, as an opensource software tool, JMod can serve as a community platform for developing new computational workflows and capabilities.
Publications
JMod: Joint modeling of mass spectra for empowering multiplexed DIA proteomics