Fantastic Proteins and Where to Find Them
Here, we will offer a tutorial for how to build instrument-specific spectral library predictions for PSMtag modified peptides using DIA-NN 2.3.0. This will allow users to do more accurate proteome-wide searches of their PSMtag data and potentially by-pass the need to first generate empirical spectral libraries from bulk or fractionated samples.
TL;DR: We've made some models so now you can predict libraries for PSMtag'd peptides. Some are here but you can make your own. Below is how. Oodles of credit to Vadim, he rocks.
Thanks to the release of proteomics search software DIA-NN 2.3.0 by Prof. Vadim Demichev, researchers can now fine-tune spectral library predictions for a range of peptide modifications, including modification by mass tags like PSMtag. Models for several modifications, including the mass tag mTRAQ, have been built into earlier versions of DIA-NN. Another proteomics search software, AlphaPeptDeep by the Mann lab, has allowed for fine-tuning spectral library predictions of modified peptides, as well. Here, we will offer a tutorial for how to build instrument-specific spectral library predictions for PSMtag modified peptides using DIA-NN 2.3.0. This will allow users to do more accurate proteome-wide searches of their PSMtag data and potentially by-pass the need to first generate empirical spectral libraries from bulk or fractionated samples. Prof. Demichev has noted that building fragmentation models is not without the pit-falls common to any model-building process, such as overtraining and not properly controlling false discovery rate.
Tutorial
Note 1: When DIA-NN opens a new window, a set of default options are loaded. In this tutorial we err on the side of changing as few of these default options as needed, since DIA-NN will turn many of them off automatically if they are not necessary for a given step. For example, we leave MBR checked, but it will automatically turn off for steps involving none or one raw file.
Note 2: If your PSMtag data is multiplexed, add the appropriate “--channels …” command with the appropriate channel masses to Steps 3 and 6.
Additional options for Steps 3 and 6 if data is 5-plexDIA:--channels tag,d0,nK,0:0; tag,d4,nK,4.01096:4.01096; tag,d8,nK,8.026839:8.026839; tag,d12,nK,12.033938:12.033938; tag,d16,nK,16.038574:16.038574
--channel-spec-normStep 0: Acquire PSMtag'd proteomic data by data-independent acquisition (DIA) on your instrument of choice. We offer the peptide-labeling protocol and suggest LC-MS methods for the Thermofisher Astral and Bruker timsTOF platforms.
Step 1: Generate a predicted spectral library using the default (label-free) models and your fasta of choice.
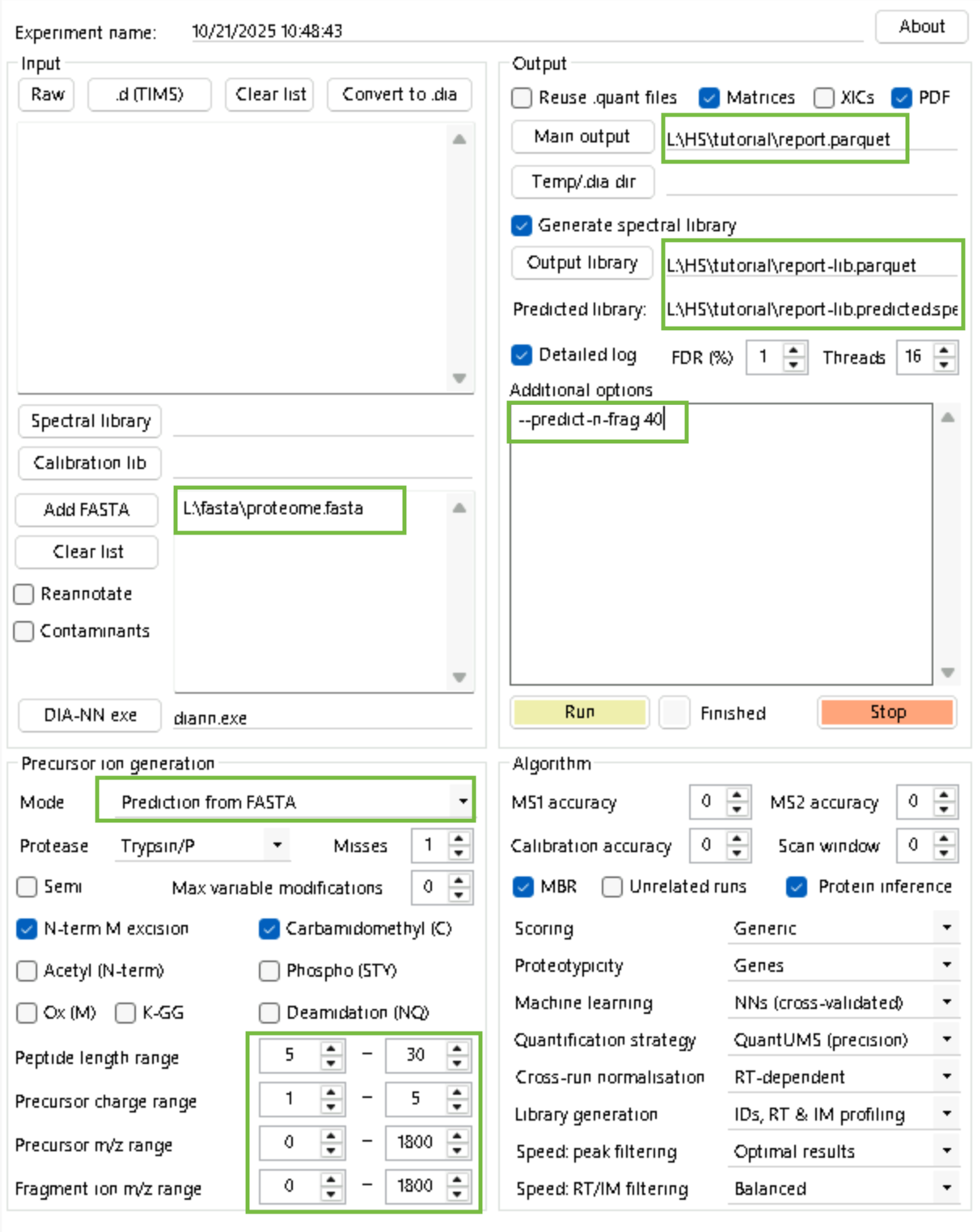
diann.exe --lib "" --threads 16 --verbose 3 --out "L:\HS\tutorial\report.parquet" --qvalue 0.01 --matrices --out-lib "L:\HS\tutorial\report-lib.parquet" --gen-spec-lib --predictor --fasta "L:\fasta\proteome.fasta" --fasta-search --met-excision --min-pep-len 7 --max-pep-len 30 --min-pr-mz 300 --max-pr-mz 1800 --min-pr-charge 1 --max-pr-charge 4 --min-fr-mz 200 --max-fr-mz 1800 --cut K*,R* --missed-cleavages 1 --unimod4 --reanalyse --rt-profilingStep 2: Add PSMtag modification to the library.

diann.exe --lib "L:\HS\tutorial\report-lib.predicted.speclib" --threads 16 --verbose 3 --out "L:\HS\tutorial\report.parquet" --qvalue 0.01 --matrices --out-lib "L:\HS\tutorial\report-lib.parquet" --gen-spec-lib --unimod4 --reanalyse --rt-profiling --fixed-mod tag, 308.1160923903, nK
--lib-fixed-mod tagStep 3 (this step may take a while): Search your data with relaxed-constraints.
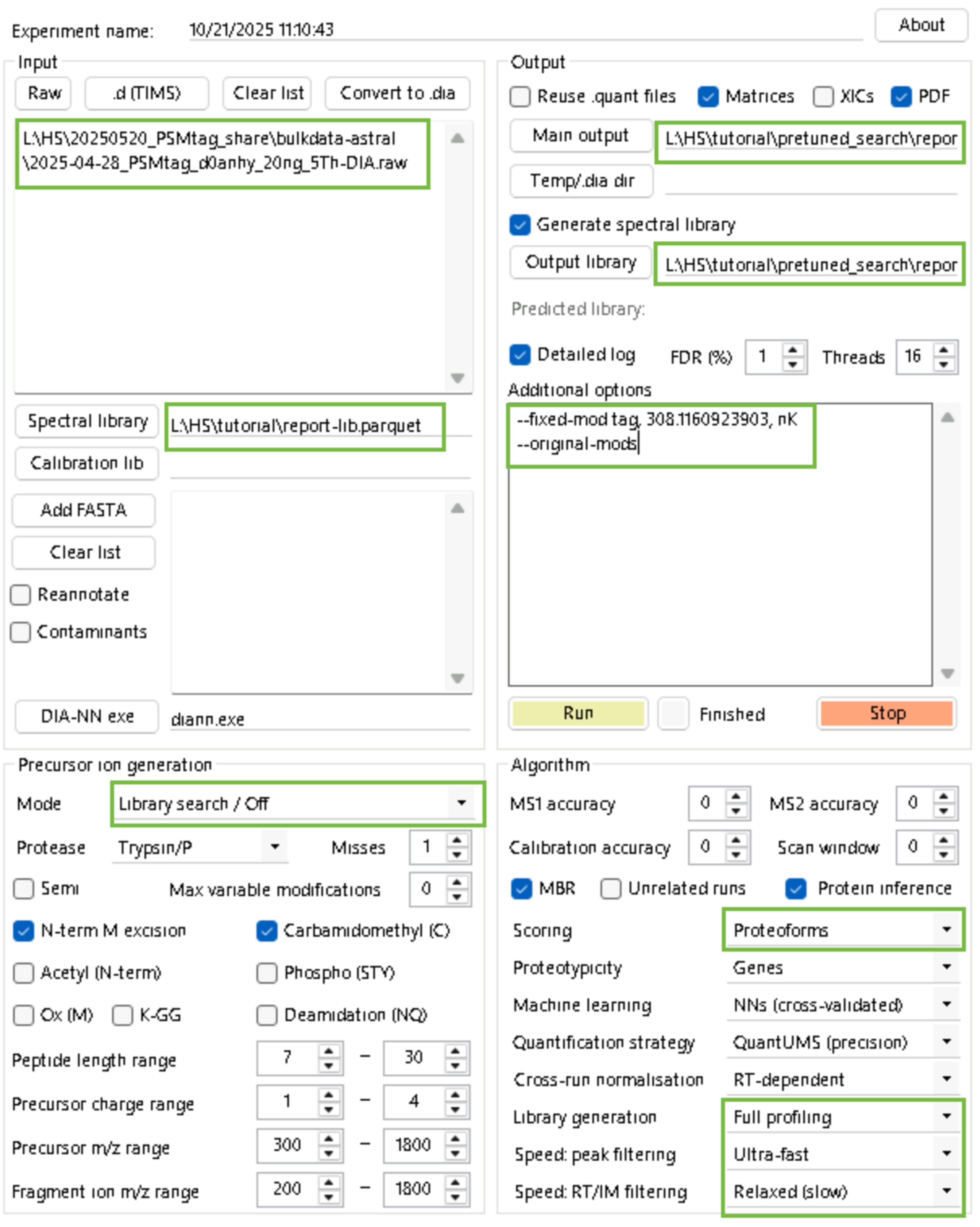
diann.exe --f "L:\HS\20250520_PSMtag_share\bulkdata-astral\2025-04-28_PSMtag_d0anhy_20ng_5Th-DIA.raw
" --lib "L:\HS\tutorial\report-lib.parquet" --threads 16 --verbose 3 --out "L:\HS\tutorial\pretuned_search\report.parquet" --qvalue 0.01 --matrices --min-corr 2.0 --time-corr-only --extracted-ms1 --min-cal 500 --min-class 1000 --pre-filter --no-rt-window --out-lib "L:\HS\tutorial\pretuned_search\report-lib.parquet" --gen-spec-lib --unimod4 --proteoforms --reanalyse --fixed-mod tag, 308.1160923903, nK
--original-modsStep 4: Train fragment, retention time and ion mobility (optional) models.
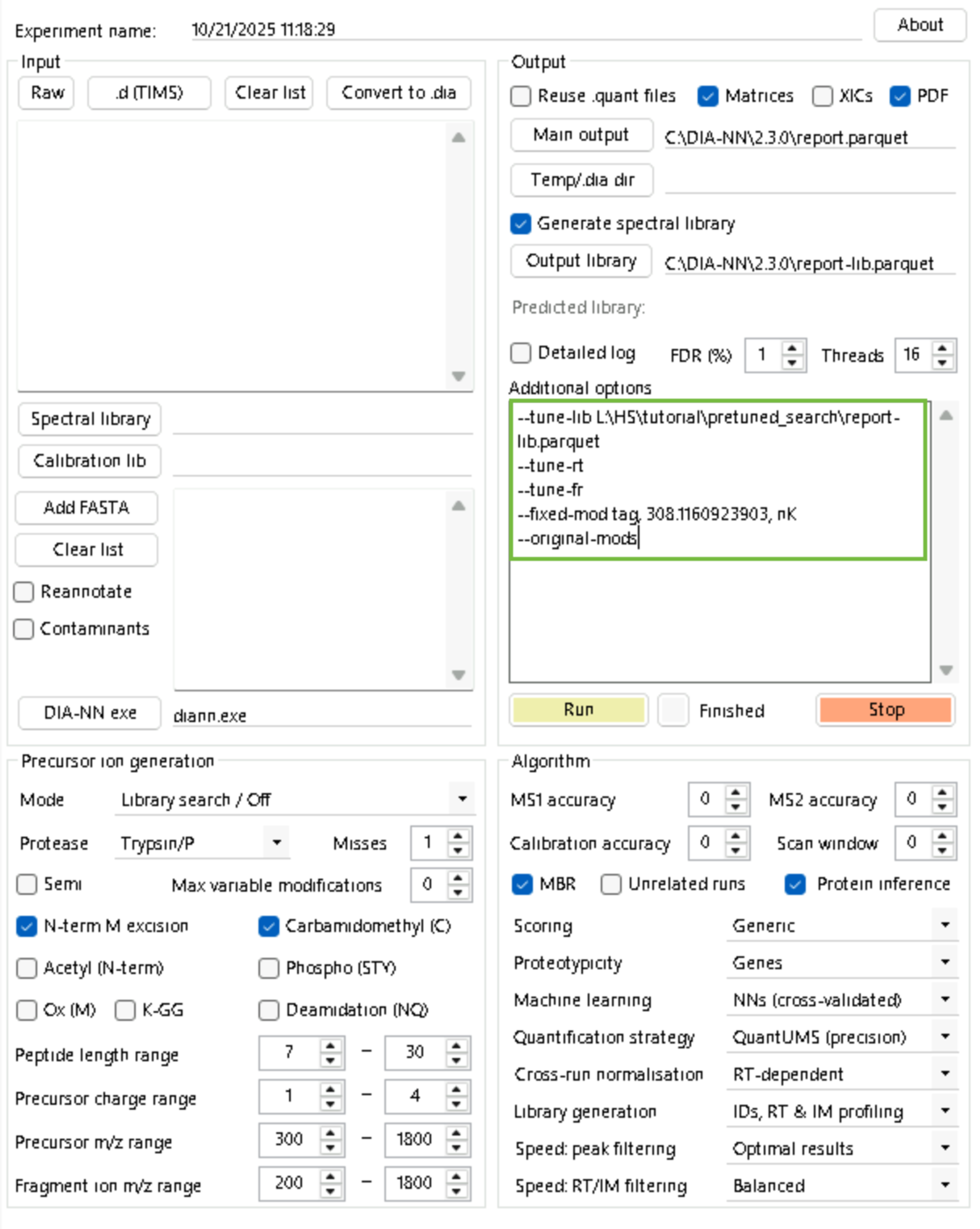
diann.exe --lib "" --threads 16 --verbose 1 --out "C:\DIA-NN\2.3.0\report.parquet" --qvalue 0.01 --matrices --out-lib "C:\DIA-NN\2.3.0\report-lib.parquet" --gen-spec-lib --unimod4 --reanalyse --rt-profiling --tune-lib L:\HS\tutorial\pretuned_search\report-lib.parquet --tune-rt --tune-fr --fixed-mod tag, 308.1160923903, nK --original-modsStep 5: Tune the library generated in Step 2 with the models generated in Step 4.
Note: If the models don’t seem to load, try renaming the files to remove the “-” and extra “.”
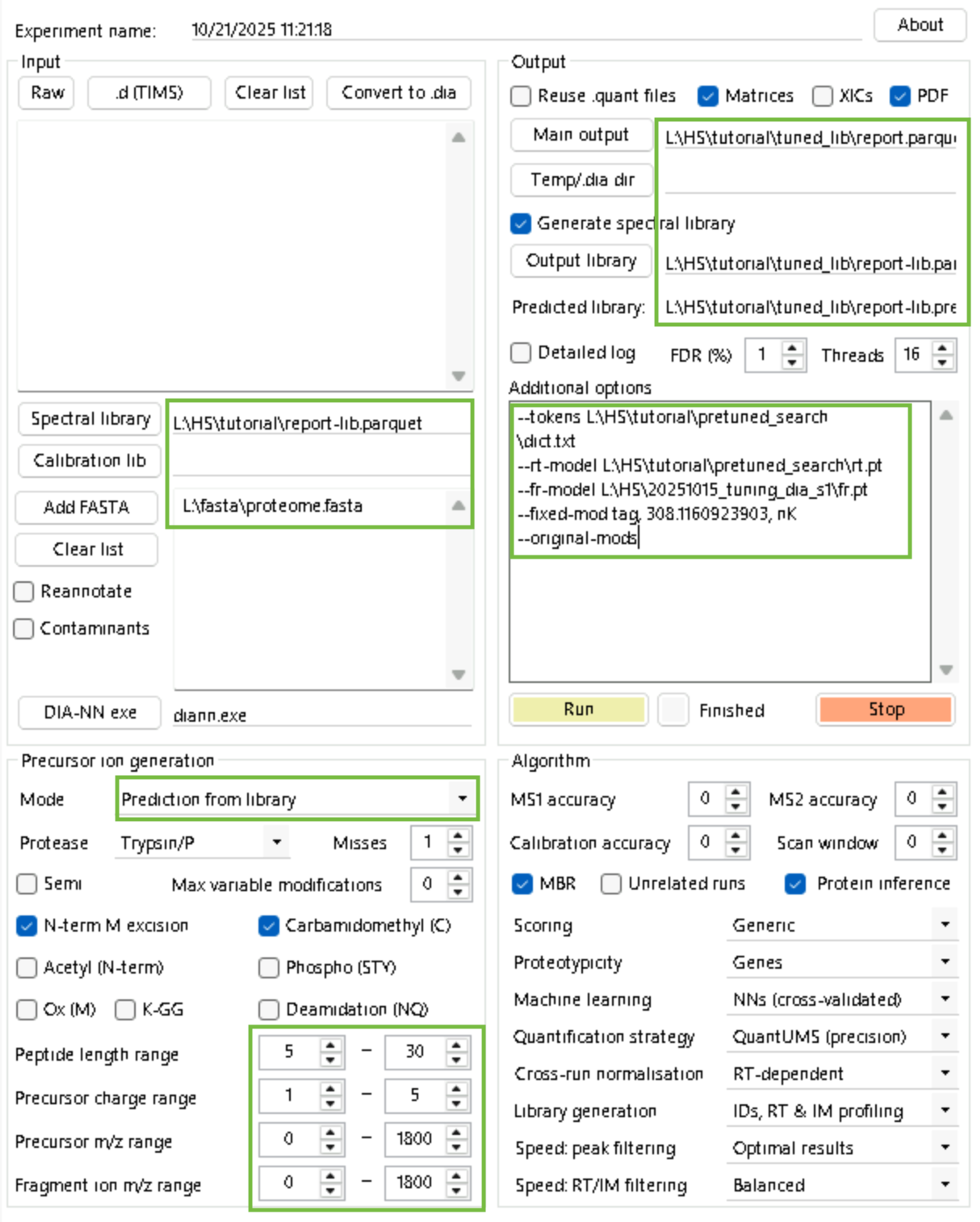
diann.exe --lib "L:\HS\tutorial\report-lib.parquet" --threads 16 --verbose 1 --out "L:\HS\tutorial\tuned_lib\report.parquet" --qvalue 0.01 --matrices --out-lib "L:\HS\tutorial\tuned_lib\report-lib.parquet" --gen-spec-lib --predictor --fasta "L:\fasta\proteome.fasta" --met-excision --min-pep-len 5 --max-pep-len 30 --min-pr-mz 0 --max-pr-mz 1800 --min-pr-charge 1 --max-pr-charge 5 --min-fr-mz 0 --max-fr-mz 1800 --cut K*,R* --missed-cleavages 1 --unimod4 --reanalyse --rt-profiling --tokens L:\HS\tutorial\pretuned_search\dict.txt --rt-model L:\HS\tutorial\pretuned_search\rt.pt --fr-model L:\HS\20251015_tuning_dia_s1\fr.pt --fixed-mod tag, 308.1160923903, nK --original-modsStep 6: Apply the fine-tuned library to other PSMtag data.
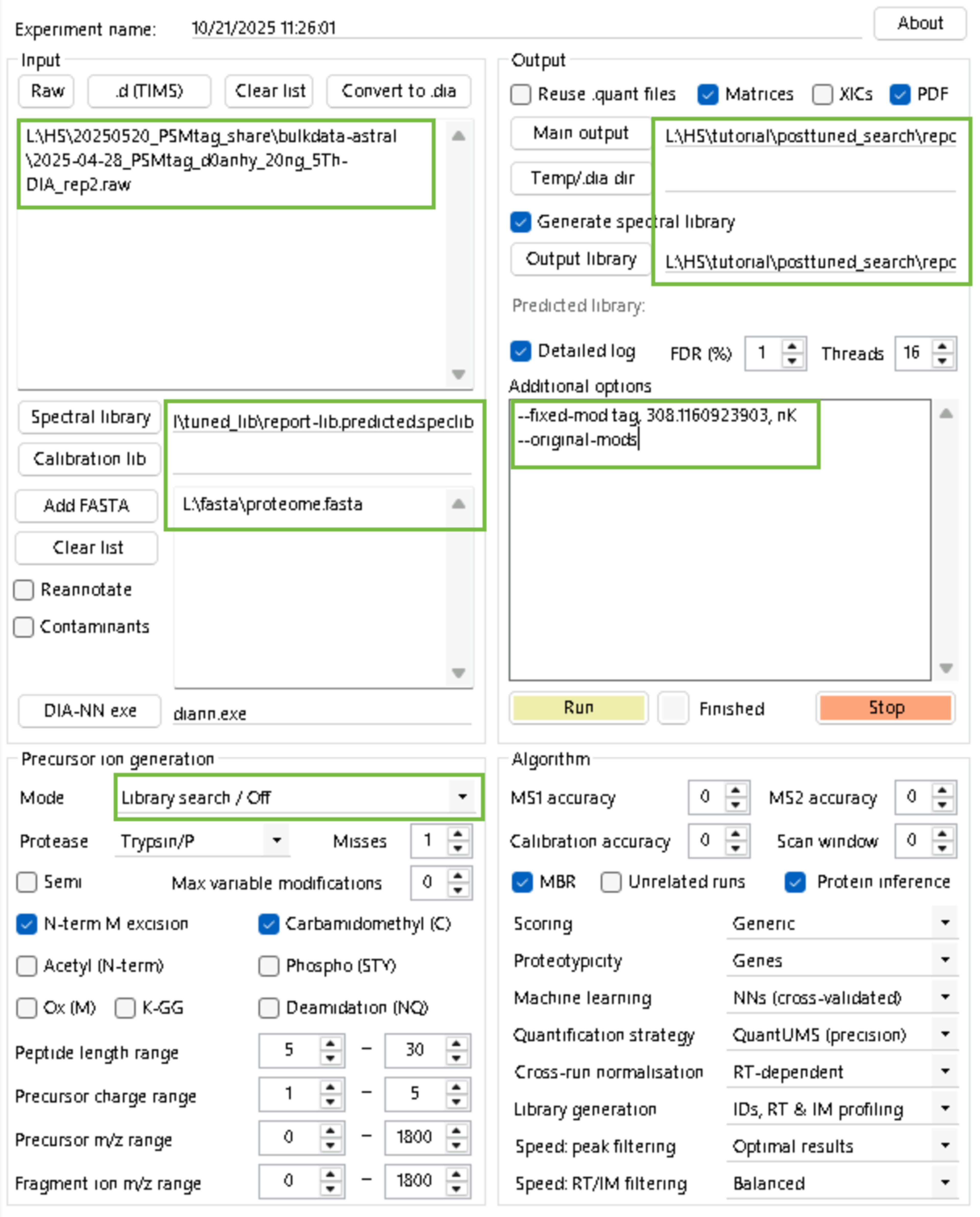
diann.exe --f "L:\HS\20250520_PSMtag_share\bulkdata-astral\2025-04-28_PSMtag_d0anhy_20ng_5Th-DIA_rep2.raw
" --lib "L:\HS\tutorial\tuned_lib\report-lib.predicted.speclib" --threads 16 --verbose 3 --out "L:\HS\tutorial\posttuned_search\report.parquet" --qvalue 0.01 --matrices --out-lib "L:\HS\tutorial\posttuned_search\report-lib.parquet" --gen-spec-lib --fasta "L:\fasta\proteome.fasta" --met-excision --min-pep-len 5 --max-pep-len 30 --min-pr-mz 0 --max-pr-mz 1800 --min-pr-charge 1 --max-pr-charge 5 --min-fr-mz 0 --max-fr-mz 1800 --cut K*,R* --missed-cleavages 1 --unimod4 --reanalyse --rt-profiling --fixed-mod tag, 308.1160923903, nK
--original-modsIf you are savvy with DIA-NN's pipeline building function, you can automate all above steps by changing the paths and filenames in the pipeline included here.