Proteomics: Greater than the sum of its parts
Over the past 2 years, PTI synergized advances in computational sciences, chemistry, biology and mass spectrometry to develop the next generation of applications and methods in single cell proteomics.

Proteins animate life. Yet, comprehensive and scalable analysis of their sequence, modifications, variation, and interactions remains limited and challenging. Even the direct determination of the primary amino acid sequence of proteomes or immunopeptidomes, is an unsolved challenge. Higher levels of complexity are more challenging still. These challenges represent a fundamental roadblock to biomedical research.
PTI seeks to clear this roadblock. We aim to increase the throughput of protein analysis while preserving or increasing coverage depth, specificity, quantitative accuracy and versatility towards functional measurements. These aims are motivated by our focus on accelerating the understanding of healthy physiology and its disruption by protein pathologies, such as neurodegenerative diseases and cancer. We embrace the challenges inherent in the proteome complexity and seek to overcome them by building scalable proteomic technologies. Our approach relies on parallelizing data acquisition and requires the simultaneous development of new chemistries, new data acquisition approaches, and data analysis software able to interpret the highly parallel measurements of our technologies; it requires a highly coordinated development of synergistic technologies, which have resulted in our first generation tools and results reported in 4 research articles published today at bioRxiv.
Parallel encoding
At a high-level, we developed new approaches and tools to parallelize the acquisition of sensitive proteomic measurements from multiple samples along two dimensions:
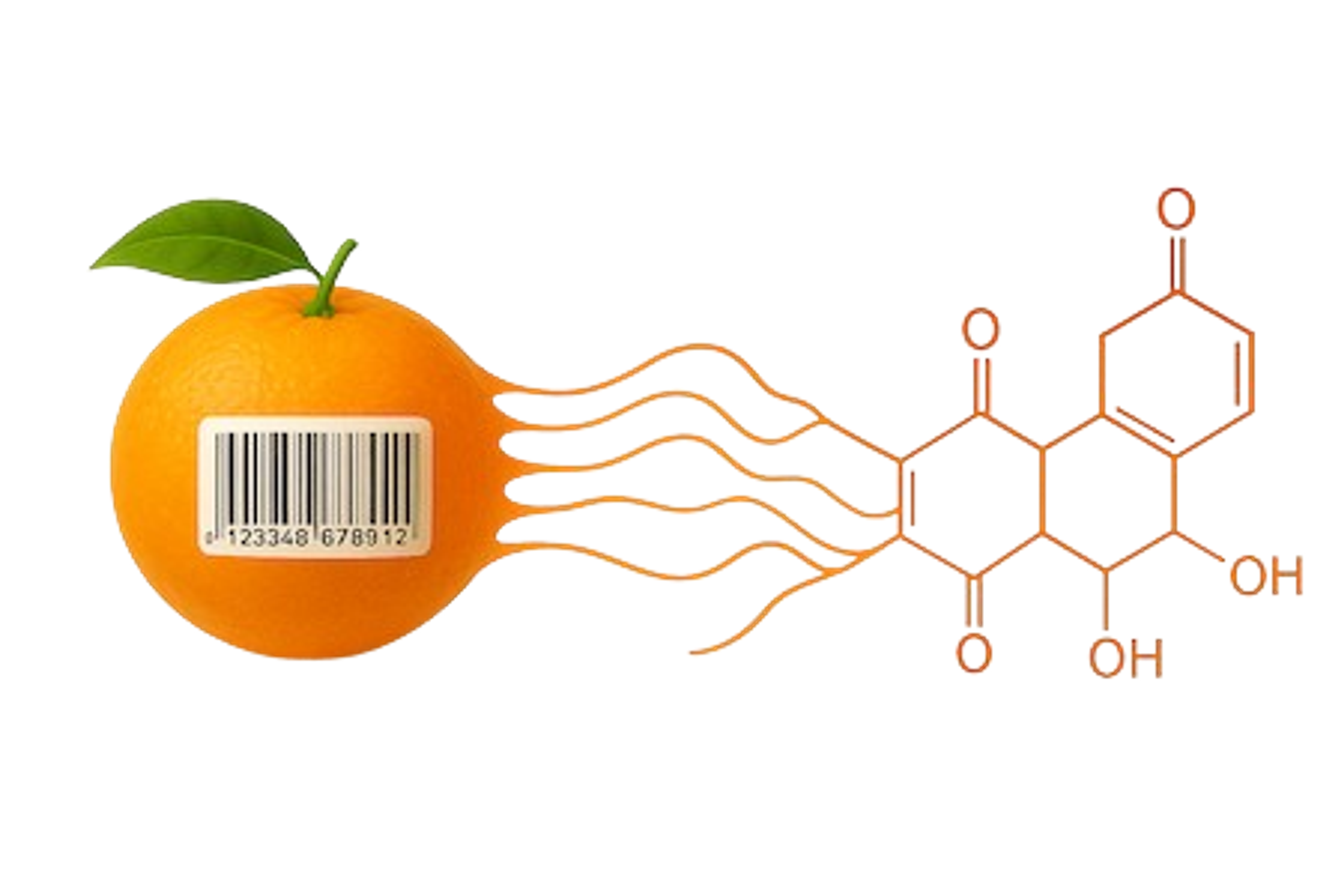
1. Parallelizing samples in the mass/charge dimension using new mass tags: PSMtags. PSMtags were developed through collaboration with MIT and CreaGen and allow for improved de novo sequencing, sensitive sequence identification, and increased multiplexing (up to 9 samples) by plexDIA. Multiplexing with PSMtags increases not only throughput but also proteomic coverage. Reported by Specht et al., 2025.
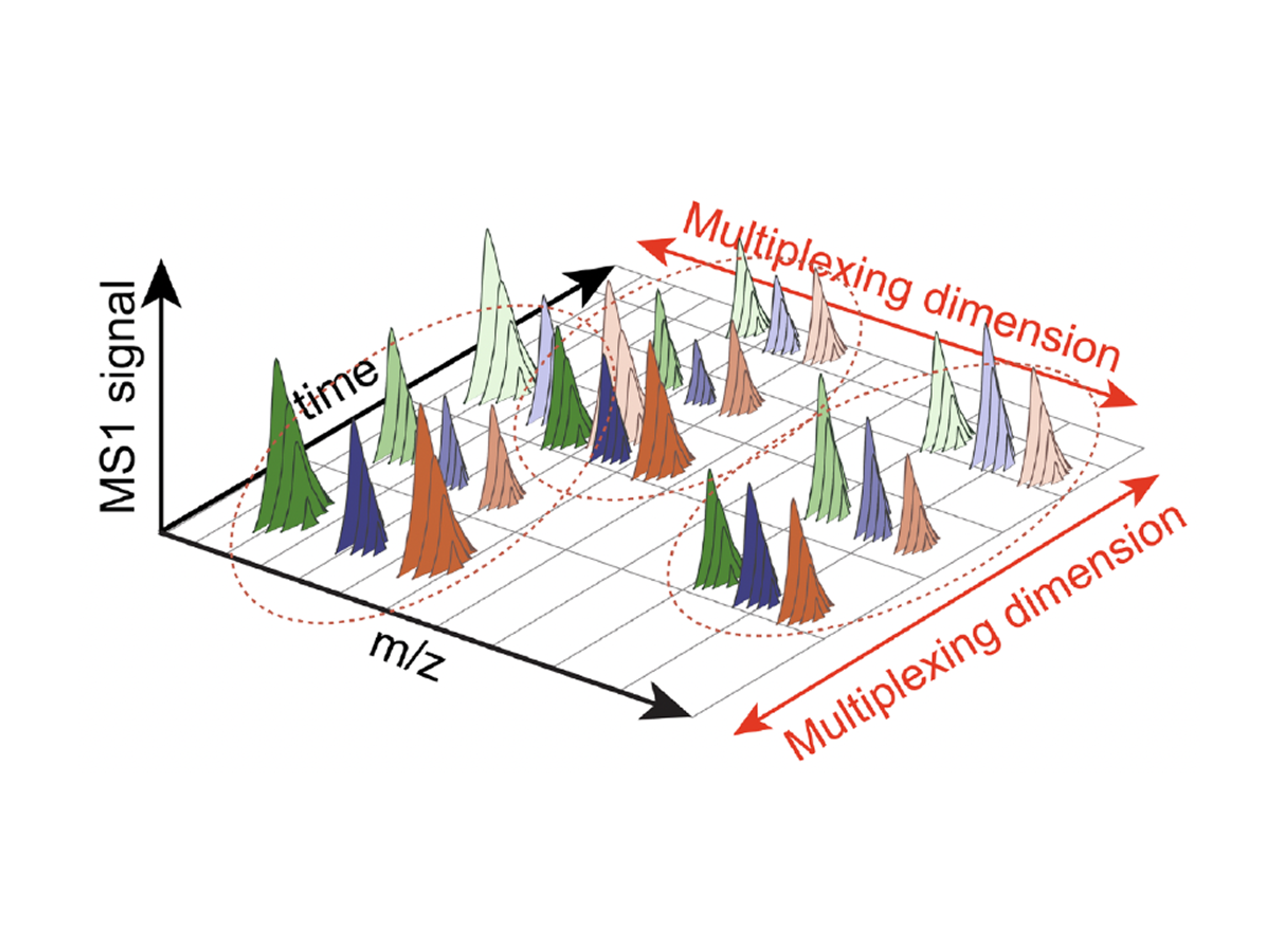
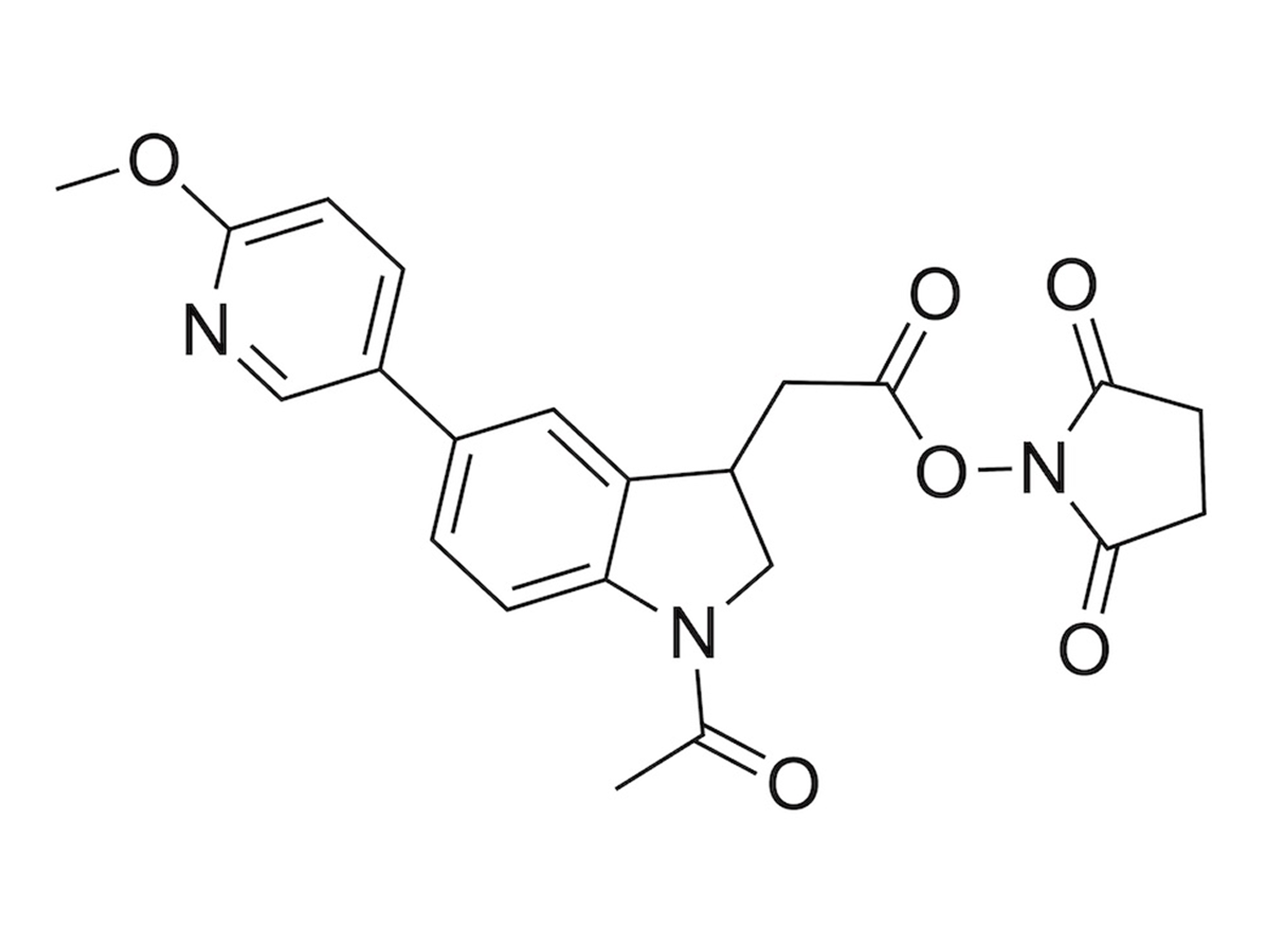
2. Parallelizing samples in the time dimension using time offsets between sample injections: timePlex. This method introduces time as an orthogonal dimension to mass for sample multiplexing, thereby allowing for combinatorial multiplexing. We demonstrate 3-timePlex combined with 9-plexDIA realizing 27-plex. This allows analyzing 27 samples per run and supports the DIA analysis of over 500 samples per day. The scaling enabled by this combinatorial multiplexing has the capacity to rapidly enable further increases beyond 1,000 samples per day. Reported by Derks et al., 2025.
Joint decoding
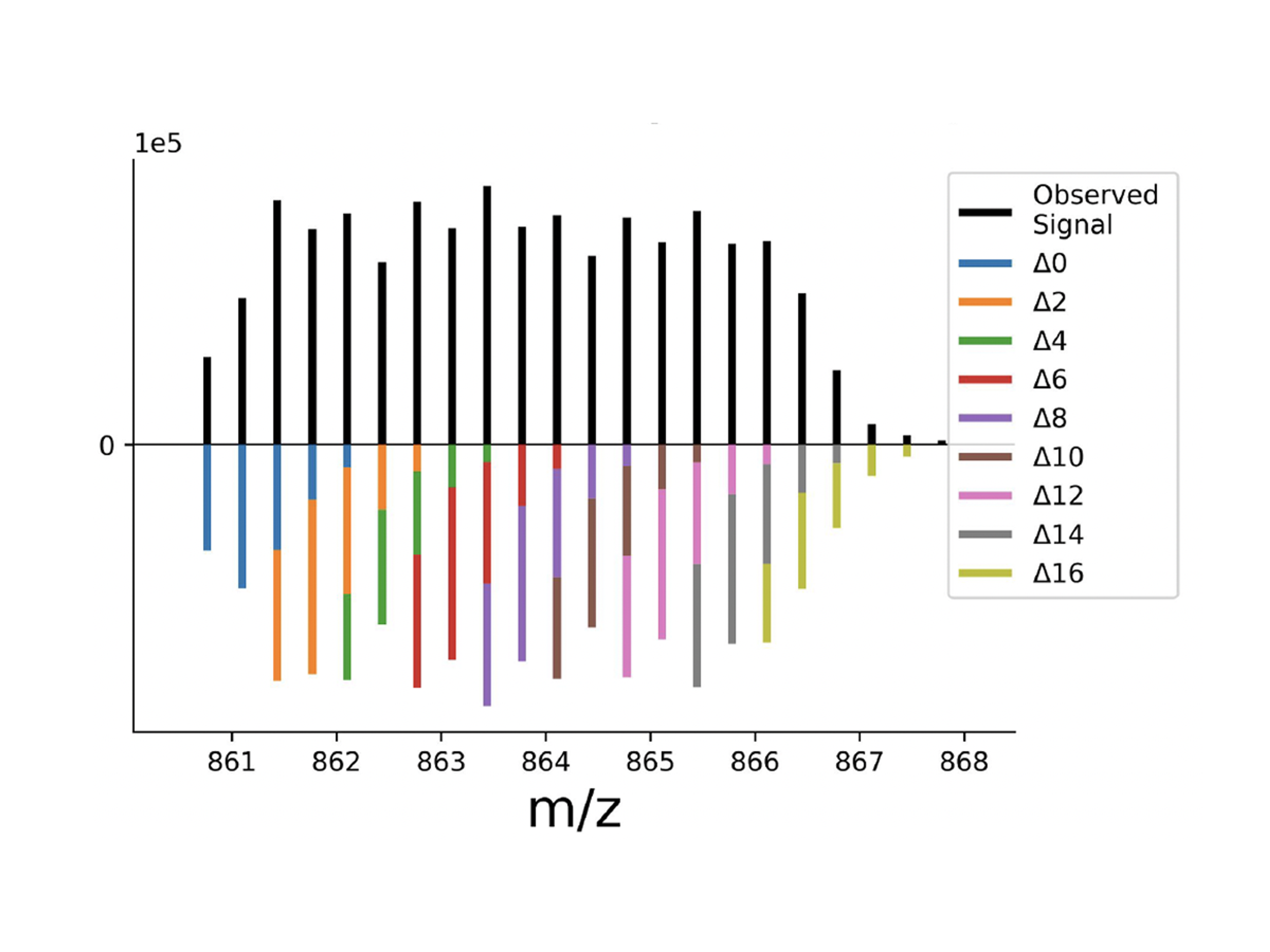
Highly parallel data acquisition generates complex mass spectra whose analysis is enhanced by our open source software for joint modeling: JMod. It uses the intrinsic structure in the spectra and explicitly models overlapping peaks as linear superpositions of their components. By supporting enhanced decoding of DIA spectra multiplexed in the mass and time domains, JMod provides an open and flexible software for increasing the throughput of sensitive proteomics. Reported by McDonnell et al., 2025.
PSMtags, timePlex and JMod are synergistic tools that support each other in scaling up the acquisition of highly specific, accurate and precise proteomic measurements. They point towards directions and methods that can continue to empower proteomic analysis. For example, the systematic framework used to identify PSMtags is now being expanded to identify new mass tags with improved performance. Similarly, new timePlex implementations are being developed to support higher plex and robustness that will advance this new direction for sample multiplexing. We are excited to share these tools with the community and thus enable more scalable protein analysis with the specificity and accuracy that mass spectrometry is known for.
Protein degradation dysregulation in Alzheimer's disease
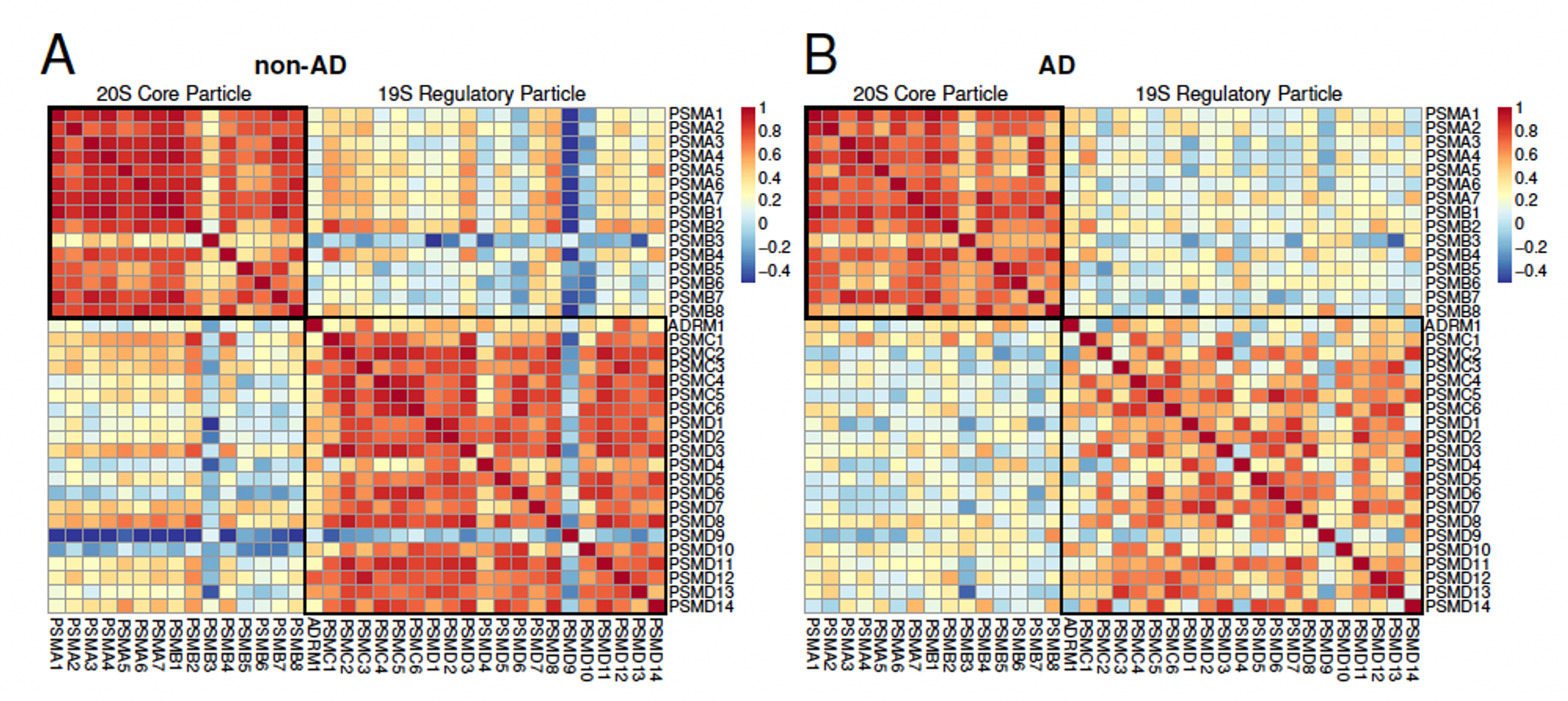
Alzheimer's disease (AD) is a relentlessly progressive, fatal neurodegenerative disorder that results in pathological protein aggregation throughout the brain. Our multiplexed protein analysis of the brain cortex from AD cases and controls identified numerous dysregulated proteins. The most prominent change in AD patients highlighted by our data is decreased abundance and coordination among the proteins making up the 20S proteasome, a protein complex required for protein degradation and quality control. Its dysfunction can further explain other protein changes associated with AD. These results emphasize the role of protein quality control in AD and point to specific molecular mechanisms likely to contribute to AD pathology. Reported by Collins et al., 2025.